-v5.svg)
-v5.svg)
Understanding Fabry Disease: A Resource Hub for Healthcare Professionals
MEDICAL SPECIALISTS




About This Resource Hub
Fabry disease is a rare, inherited disorder that can have a significant impact on multiple organ systems.
This page is designed to provide healthcare professionals with essential information, educational resources, and tools to better understand and manage Fabry disease.
Stay informed, stay educated, and together, we can improve the management of this rare condition.
What is Fabry disease?
Fabry disease is a genetic disorder caused by a deficiency in the enzyme alpha-galactosidase A, leading to the accumulation of a type of fat called globotriaosylceramide (GL-3) in the body's cells.
This buildup affects various organs, including the kidneys, heart, and nervous system, leading to a wide range of symptoms.
Patients may present with symptoms such as pain in the hands and feet, gastrointestinal issues, skin rashes, and more severe complications like kidney failure or heart disease.
What is Fabry Disease?
This buildup affects various organs, including the kidneys, heart, and nervous system, leading to a wide range of symptoms.
Patients may present with symptoms such as pain in the hands and feet, gastrointestinal issues, skin rashes, and more severe complications like kidney failure or heart disease.
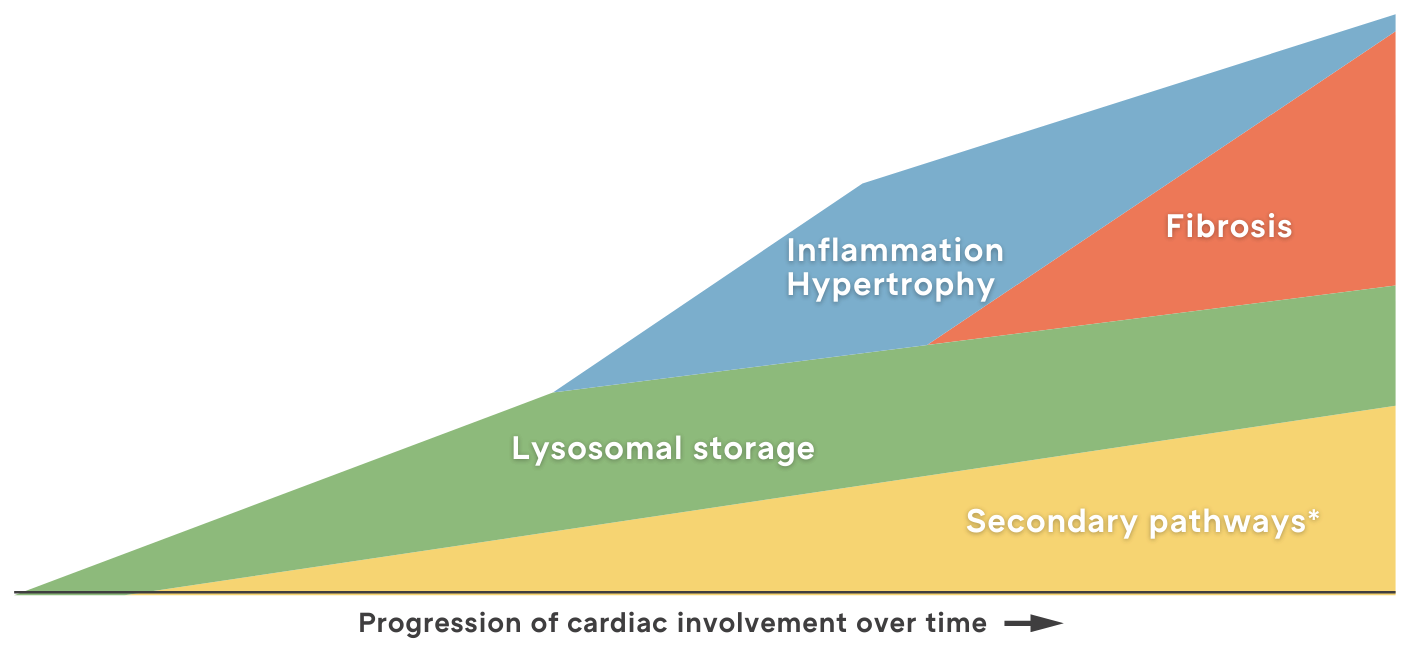
Proposed evolution of Fabry disease
In Fabry Disease, Early Diagnosis Leads to Better Outcomes
*Refers to recently discovered mechanisms active in Fabry disease including dysfunction of the endoplasmic reticulum, ion channels, Golgi bodies, mitochondria, and sarcomeres; impairment of energy production and autophagy; and apoptosis.1
Fabry disease, while often challenging to diagnose, is highly treatable when detected early. Early intervention can significantly improve patient outcomes.
Given its multisystemic nature, it is crucial to consider Fabry disease in patients with unexplained pain, kidney issues, or a family history of the disease. Prompt diagnosis and referral to a specialist are key
Fabry Disease for Cardiologists
Cardiovascular disease is the most common cause of death for both men and women with Fabry disease.2
Cardiac involvement often presents early in life during adolescence in both genders, with cardiac symptoms appearing ~10 years earlier in males than in females.4 Both sexes are affected by cardiac symptoms, with ~90% of patients being affected by cardiomyopathy.

Fabry Education For Cardiologists

Fabry Resources For Cardiologists

Fabry Disease for Nephrologists
Could Fabry disease be the cause of your patient's kidney disease? Identifying Fabry disease early is crucial before irreversible organ damage occurs.
Fabry Resources For Nephrologists
Fabry Education For Nephrologists


Fabry Disease for Neurologists
Neurologists should consider Fabry disease in patients presenting with neurological manifestations such as stroke, headaches, dizziness and pain. Keep in mind that cerebrovascular complications are the second leading cause of death in men with Fabry disease.2 Early diagnosis and treatment are essential.
As a GP, you play a crucial role in facilitating the early diagnosis of Fabry disease and providing ongoing treatment support. Given the rarity and complexity of this condition, a solid understanding of Fabry disease is invaluable in optimising patient outcomes. This section aims to equip you with the knowledge and resources necessary to effectively diagnose, refer, and support your patients.
'As Fabry disease causes tissue damage in a number of organ systems, the therapeutic approach to patients with Fabry disease should ideally be multidisciplinary and integrated into a comprehensive medical care plan that addresses individual patient health needs.' 6

Fabry Disease for General Practitioners
Fabry Resources For General Practitioners

Fabry Education For General Practitioners


Fabry Disease for Gastroenterologists
Gastrointestinal (GI) symptoms are a common, early-onset sign of Fabry disease appearing in childhood and continuing into adulthood.7 Irritable bowel syndrome is a misdiagnosis that may be given to patients with Fabry disease,1 as they may experience bouts of diarrhoea followed by periods of constipation.8

Fabry Disease for Optometrists & Opthalmologists
Fabry Disease is a rare disease that does not typically disturb visual function. However, ocular signs of corneal verticilata, spoke-like cataract, or vessel tortuosity may be key to its diagnosis.
The variable nature of the symptoms and signs means that diagnosis may be delayed further, emphasising the role of the ophthalmologist in assisting with identification of this group of patients.
Fabry Resources For Ophthalmologists
Fabry Education For Ophthalmologists

Fabry Disease for Geneticists
Neurologists should consider Fabry disease in patients presenting with neurological manifestations such as stroke, headaches, dizziness and pain. Keep in mind that cerebrovascular complications are the second leading cause of death in men with Fabry disease. Early diagnosis and treatment are essential.
Fabry Education For Geneticists
Fabry Resources For Geneticists
FAQs for All Medical Professionals
What are the clinical signs and symptoms of Fabry disease?
Fabry disease presents with diverse symptoms, including neuropathic pain, angiokeratomas, hypohidrosis, gastrointestinal discomfort, renal dysfunction, and cardiovascular complications. Symptoms vary in severity and may be misattributed to more common conditions.
What are the challenges in diagnosing Fabry disease in clinical practice?
Diagnosing Fabry disease can be challenging due to its rarity, variable presentation, and overlap with other conditions. Genetic testing and awareness of red-flag symptoms are critical for timely diagnosis.
How does Fabry disease impact multi-organ systems?
Fabry disease affects multiple systems, including the cardiovascular, renal, neurological, and gastrointestinal systems, through the accumulation of globotriaosylceramide (Gb3) in cells, leading to progressive damage over time.
What are the current guidelines for managing Fabry disease in adults?
Management guidelines emphasise early diagnosis, initiation of enzyme replacement therapy or chaperone therapy, regular monitoring of organ function, and multidisciplinary care to address complications.
How can healthcare professionals identify at-risk patients for Fabry disease?
At-risk patients include those with unexplained left ventricular hypertrophy, stroke at a young age, renal dysfunction, or a family history of Fabry disease. Screening and genetic testing are key.
Fabry FAQs for Cardiologists
How does Fabry disease affect the cardiovascular system?
Fabry disease can cause left ventricular hypertrophy, arrhythmias, valve dysfunction, and heart failure due to Gb3 accumulation in myocardial cells and vascular endothelial cells.
What cardiac imaging techniques are recommended for patients with suspected Fabry disease?
Cardiac MRI with late gadolinium enhancement and echocardiography are valuable tools for identifying characteristic features of Fabry cardiomyopathy, such as left ventricular hypertrophy and fibrosis.
What are the signs of left ventricular hypertrophy associated with Fabry disease?
Left ventricular hypertrophy in Fabry disease often presents with symmetrical thickening and may be accompanied by diastolic dysfunction and arrhythmias, which can mimic hypertrophic cardiomyopathy.
Fabry Disease FAQs for Gastroenterologists
What gastrointestinal symptoms are associated with Fabry disease?
Patients with Fabry disease may experience abdominal pain, diarrhoea, bloating, and nausea, often resulting from Gb3 accumulation in the gastrointestinal tract and autonomic nervous system.
How can Fabry disease mimic other gastrointestinal disorders?
Symptoms from irritable bowel syndrome, inflammatory bowel disease, or functional dyspepsia can overlap with Fabry-related gastrointestinal issues, leading to misdiagnosis without genetic testing.
What role does enzyme replacement therapy play in managing gastrointestinal symptoms?
Enzyme replacement therapy can reduce Gb3 accumulation and improve gastrointestinal symptoms in patients with Fabry disease, contributing to overall quality of life.
Fabry Disease FAQs for General Practice
What is the role of the GP in diagnosing and managing Fabry disease?
GPs play a vital role in identifying early signs, initiating genetic testing, and coordinating care with specialists to manage Fabry disease and its complications effectively.
When should a GP refer a patient with suspected Fabry disease to a specialist?
Referral is recommended for patients with red-flag symptoms (e.g., unexplained kidney issues, cardiac symptoms, or neuropathic pain) or a positive family history of Fabry disease.
What are the red flags for Fabry disease in primary care?
Red flags include unexplained pain, proteinuria, premature stroke, left ventricular hypertrophy, cornea verticillata, and a family history of Fabry disease.
Fabry Disease FAQs for Geneticists
What genetic mutations are associated with Fabry disease?
Fabry disease is caused by mutations in the GLA gene, which encodes the enzyme alpha-galactosidase A. Over 1,000 pathological mutations have been identified, with varying clinical implications.
How does Fabry disease inheritance work (X-linked inheritance)?
Fabry disease follows X-linked inheritance. While males often experience more severe symptoms, females can also develop significant manifestations due to random X-chromosome inactivation.
What is the importance of genetic counselling for families affected by Fabry disease?
Genetic counselling helps families understand the inheritance pattern, assess the risk for relatives, and facilitate early diagnosis and intervention.
Fabry Disease FAQs for Nephrologists
How does Fabry disease lead to renal complications?
Gb3 accumulation in renal cells causes glomerular, tubular, and vascular damage, leading to proteinuria, reduced glomerular filtration rate, and eventually end-stage kidney disease.
What are the key biomarkers for detecting Fabry nephropathy?
Key biomarkers include proteinuria, reduced alpha-galactosidase A enzyme activity, elevated plasma lyso-Gb3 levels, and evidence of Gb3 accumulation on kidney biopsy.
What treatment options are available for Fabry-related kidney disease?
Enzyme replacement therapy and chaperone therapy slow renal progression, while adjunct treatments like ACE inhibitors or ARBs manage proteinuria and hypertension.
Fabry Disease FAQs for Neurologists
What neurological symptoms are indicative of Fabry disease?
Neurological symptoms include burning pain in hands and feet (acroparesthesia), dizziness, headaches, and increased risk of stroke or transient ischemic attacks at a young age.
How does Fabry disease contribute to cerebrovascular events like stroke?
Fabry disease causes small vessel disease due to Gb3 deposition in cerebral blood vessels, increasing the risk of ischemic strokes, particularly in younger patients.
What tools can neurologists use to diagnose neuropathy in Fabry disease?
Neurologists can use nerve conduction studies, quantitative sensory testing, and assessment of lyso-Gb3 levels to diagnose and monitor Fabry-related neuropathy.
Fabry Disease FAQs for Ophthalmologists
What ocular manifestations are commonly seen in Fabry disease?
Cornea verticillata (whorled opacity) is a hallmark feature. Other findings include conjunctival vessel tortuosity and posterior capsular cataracts.
How can cornea verticillata be used as a diagnostic clue for Fabry disease?
Cornea verticillata is easily observed during slit-lamp examination and is often an early, non-invasive indicator of Fabry disease.
What is the role of routine eye exams in monitoring Fabry disease progression?
Routine eye exams can help detect early signs of Fabry disease, monitor disease progression, and provide clues for systemic involvement.
Fabry Disease Frequently Asked Questions (FAQs)
Praxhub would like to acknowledge Amicus Therapeutics for its support of this resource and education hub.
References:
- Pieroni M, Moon JC, Arbustini E, et al. Cardiac involvement in Fabry disease. J Am Coll Cardiol. 2021;77(7):922-936.
- Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. Genet Med. 2009;11(11):790-796
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30.
- Linhart A, Kampmann C, Zamorano JL, et al. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J. 2007;28(10):1228-1235.
- Kampmann C, Perrin A, Beck M. Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: cardiac outcomes after 10 years’ treatment. Orphanet J Rare Dis. 2015;10:125.
- Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124(3):189-203.
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30.
- Hilz MJ, Arbustini E, Dagna L, et al. Non-specific gastrointestinal features: could it be Fabry disease? Dig Liver Dis. 2018;50(5):429-437.
To access hundreds of quality CPD training courses, simply join Praxhub's online medical community
© Praxhub 2026, all rights reserved.
PP-NN-AU-0001-1224